Starfish™ is an AI and physics-based structure generation pipeline enabling fast, accurate and physically plausible capsid structure generation capable of large library screening tasks. Capsids are large, complex multimers which are poorly supported by the current generation of protein folding algorithms. Starfish™ rapidly identifies relevant binding surfaces for capsid multimers using cost-efficient hardware, enabling Lir to incorporate essential capsid structural analysis into large-scale viral variant library screenings.
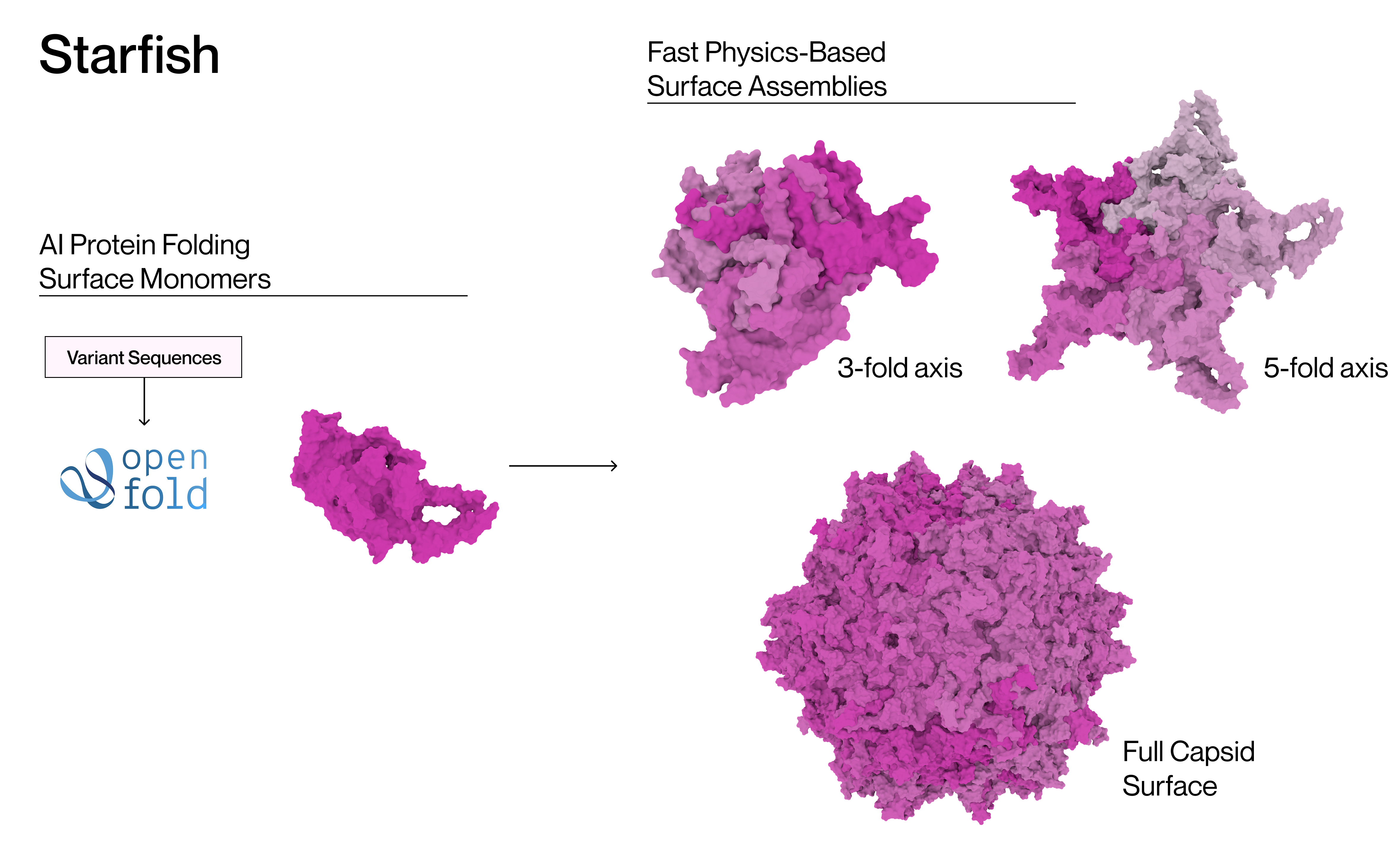
The first stage of our pipeline involves applying a protein folding model to generate the monomers involved in the capsid surface. This is followed by fast physics-based assembly and refinement algorithms. Our method scales far better than multimer cofolding both in terms of runtime and compute requirements, with Starfish™ able to assemble the full capsid at acceptable runtimes for screening tens of thousands of variants. Our approach resolves unrealistic clashes often produced by AI-generated structures, while anchoring the resulting complex in physics-based models suitable for downstream tasks such as molecular docking.
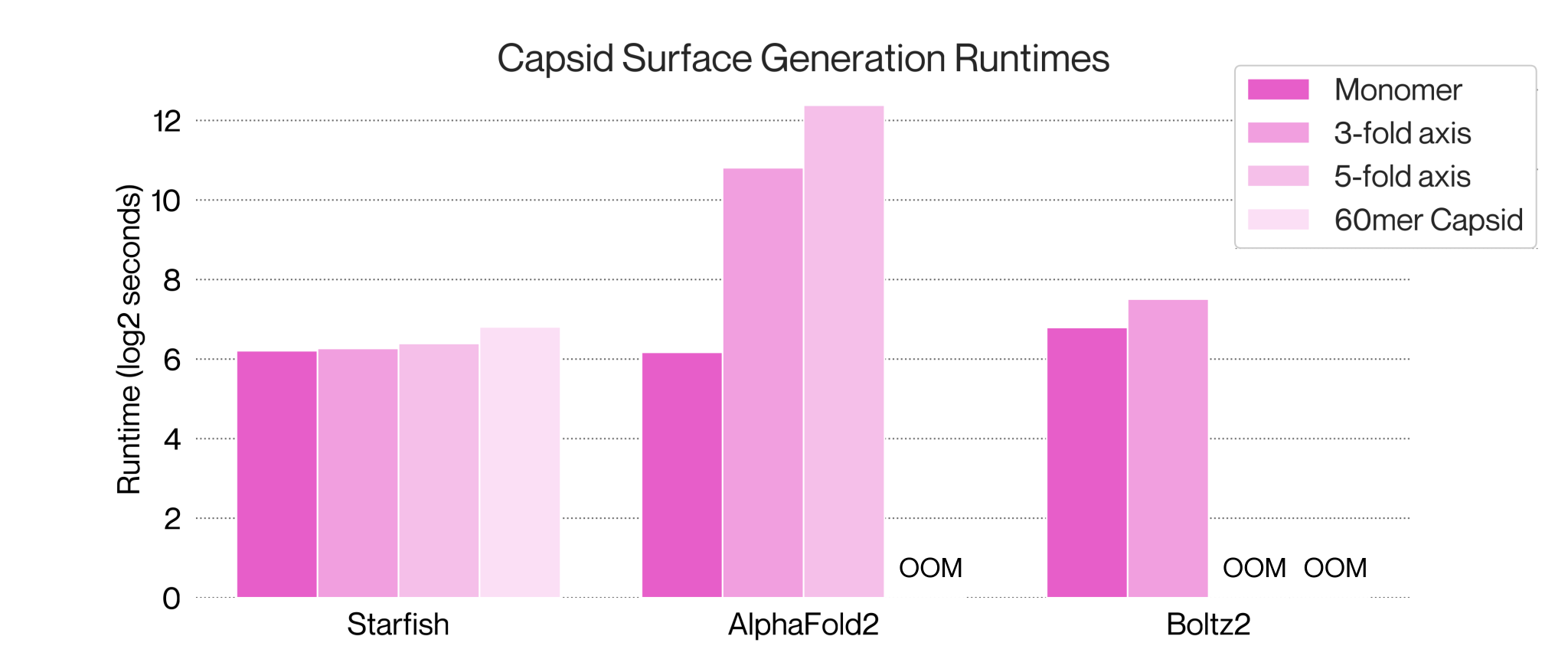
We built Starfish™ because existing models like Boltz2 and Alphafold aren’t capable of modelling whole viruses at the throughput we need.
Viral capsids are made up of multiple overlapping proteins that form the surface of the virus. Much of Lir's work involves mutating the surface of capsids to alter the properties of viruses for gene therapy. Existing protein folding models can compute the monomers of most proteins involved in viral capsids with high accuracy, but struggle to both quickly and accurately generate complete surfaces.
Compared to some other viruses, AAV has a relatively simple capsid made up for 60 subunits. Computing large portions of the capsid using models such as Alphafold or Boltz2 is impractical when scaling to a large number of targets due to both their time and memory requirements. Owing to its structural homogeneity, modelling the complete 60-mer AAV capsid is usually unnecessary, but portions of the surface such as at the 2, 3 and 5-fold axes are the minimum subunit for useful docking or structural assessment of capsid stability in silico.
All approaches generate the 3-fold axes of AAV9 wild-type with an RMSD <1Å, but Starfish™ provides a clear advantage in runtime and scalability. At the 5-fold axis Boltz2 exceeds the memory capacity of an 80GB H100 GPU to compute the structure making it cost-prohibitive for larger screening experiments. Alphafold2 Multimer eventually generates a pentameric structure but it is not the correct 5-fold axis. Alphafold3 had the same incorrect output structure in our tests. Our approach generates the correct structure efficiently, at an acceptable runtime for large screens and with no clashes for docking or analysis.

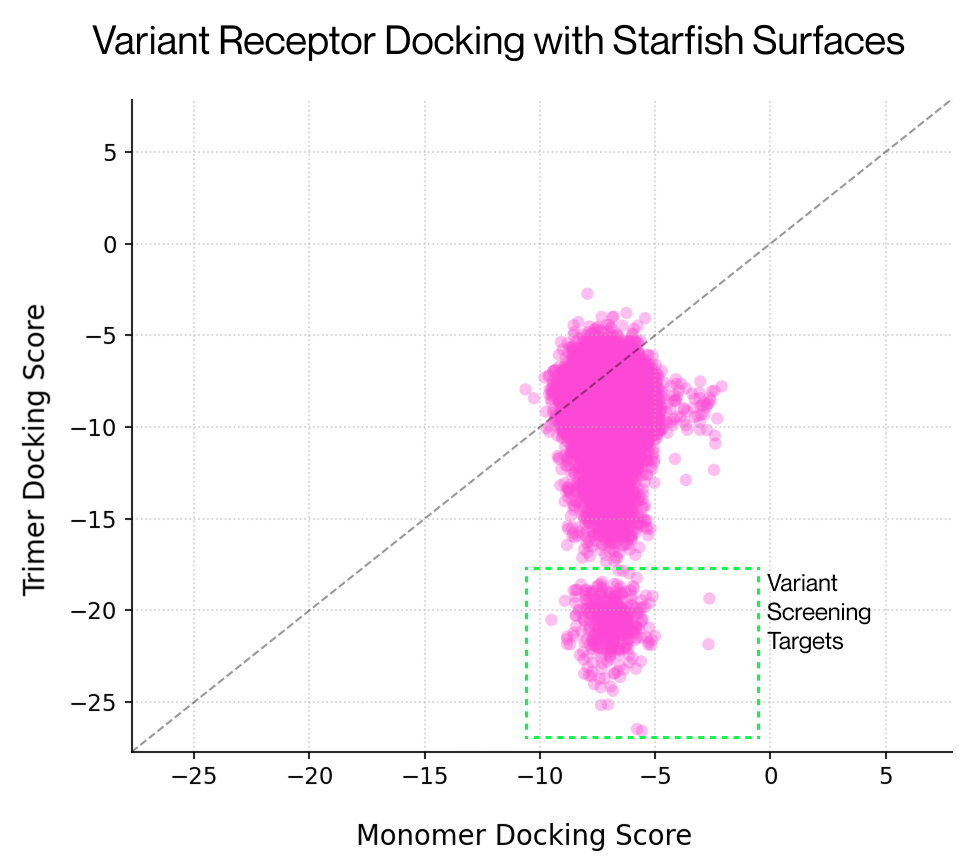
We first showcased an application of this technology at ESGCT 2025 in Seville, demonstrating that screening of the LY6A receptor was possible by performing docking to variant capsid surfaces constructed using Starfish™. We found a structural approach identified an alternative subset of hits compared to a language model-based approach, with the two methods being additive. We've been using this same approach for other receptors to investigate experimental targets predicted to exhibit differential binding. Multimer surfaces are essential to perform these larger screens, with no detectable signal present with the monomer only. For these applications Starfish™ is a crucial part of our pipeline, enabling rapid surface generation with minimal compute overheads.

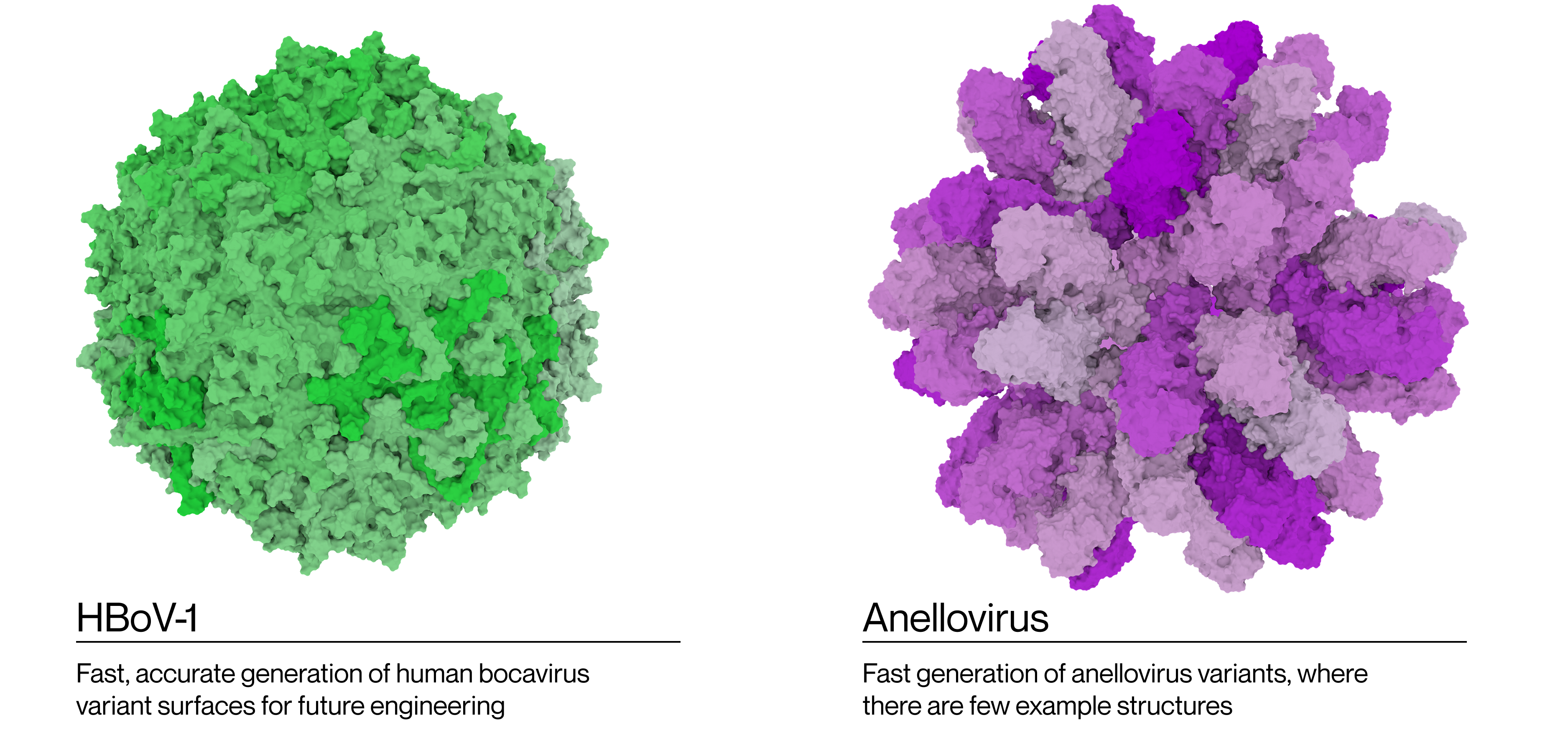
Starfish™ is also being extended to other viruses in line with Lir's ambitions to build a state-of-the-art viral engineering toolkit for gene therapy. Combined with our DNA and Protein language models for viruses, Starfish™ allows us to consider viral engineering at multiple levels of analysis.
Starfish™ is one part of Lir's nAAVigator® pipeline which is designed to accelerate viral vector design. Combined with our lab-in-the-loop, we’re building a platform to generate vectors specific to clinical partners’ targets at speed. We'll continue to post development updates here in the coming months. If this sounds relevant to your work please reach out!
© 2025 Copyright: Lir Therapeutics