LirEvolve is a large state-space model trained with a curated set of viral data, resulting in a relevant foundation model for viruses. This serves as both a zero-shot predictor for highly novel mutation targets and as a strong foundation for downstream prediction tasks on our in-house data. Lir is generating novel datasets and needed a foundation model ready to utilise them.
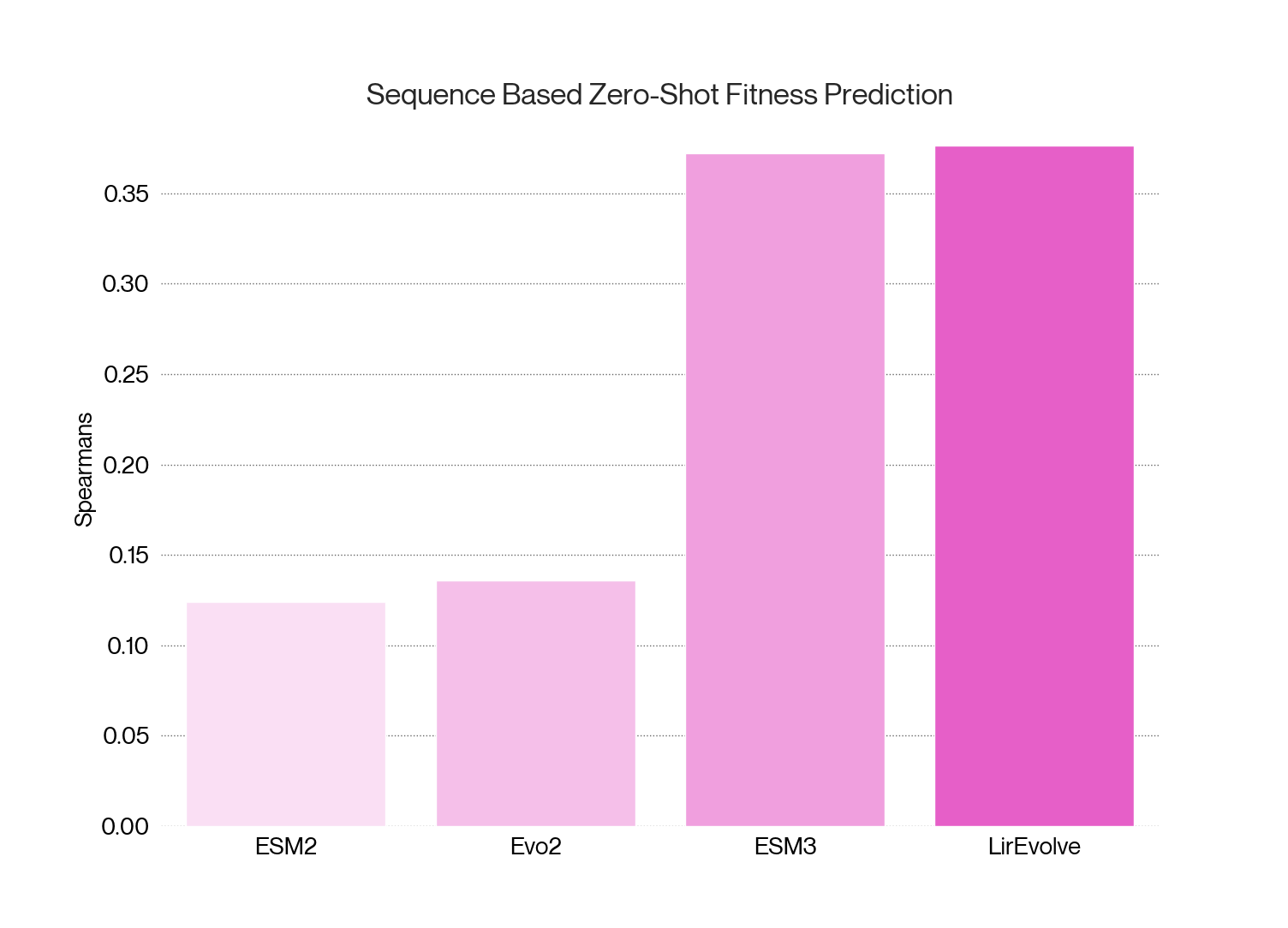
Initially we benchmarked our model on a subset of the ProteinGym dataset, which contains a relevant set of viral mutagenesis fitness data. Here we found high zero-shot performance compared to other methods using sequence inputs alone. The open license models we compared all performed relatively poorly on relevant benchmarks. ESM3 outperformed other tested models, but it is not available for open commercial use and is significantly larger than our model. LirEvolve outperformed all compared models. These results gave us the confidence to begin using LirEvolve as one step in filtering our candidates in mutable regions not covered by existing data. As we progress we continue to validate and improve LirEvolve with our in-house datasets.
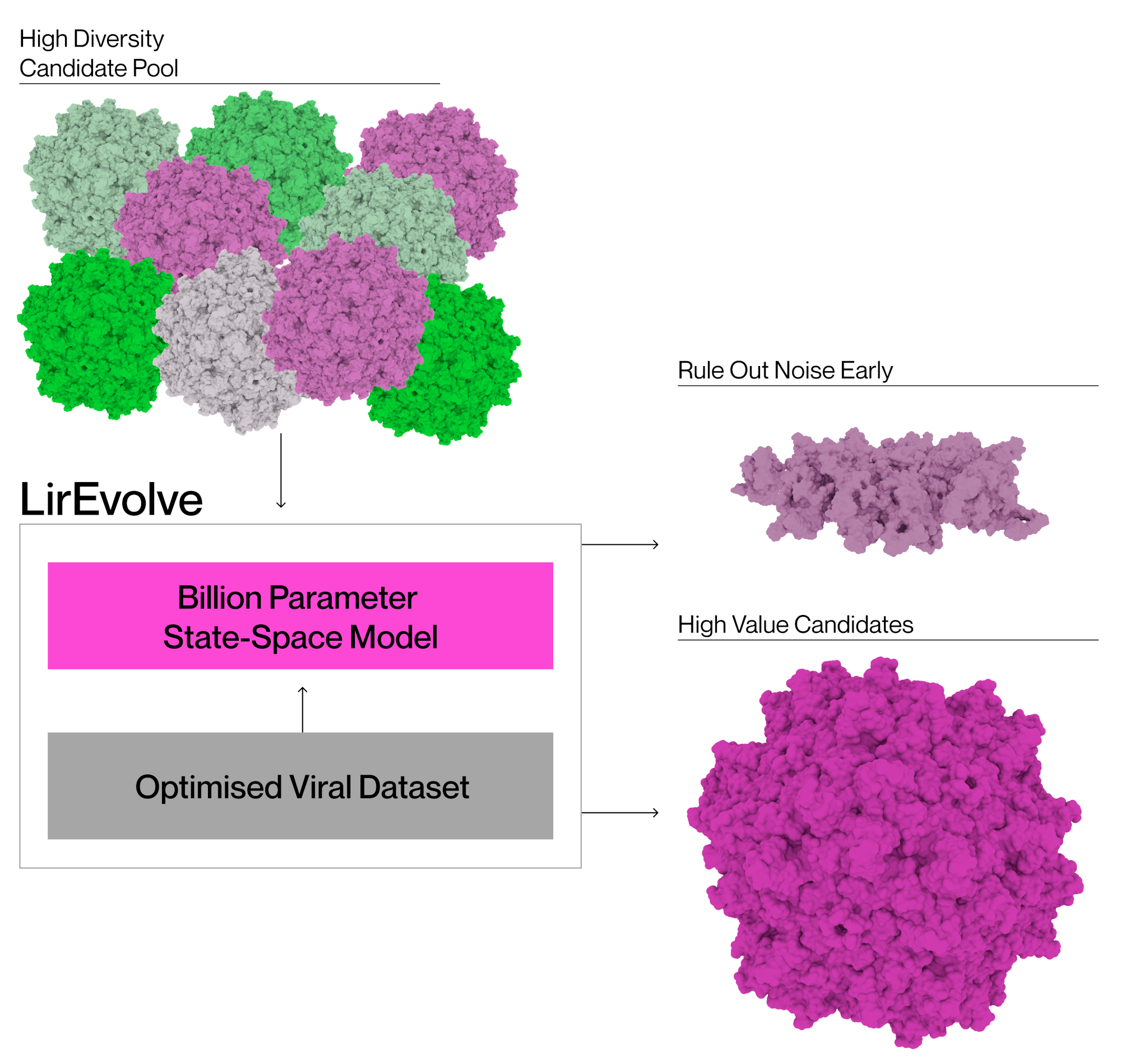
LirEvolve is a state-of-the-art DNA foundation model targeted specifically towards viruses. It was designed to perform zero/few shot prediction tasks in low data settings, and is accelerating Lir’s viral engineering work already! We built LirEvolve because fundamentally, existing models aren't up to the tasks we need to solve.
Foundation models for biology have continued to improve and expand their capabilities beyond what was once thought possible in silico. Iterations on previous models such as Alphafold3 showed it was possible to extend protein folding models to predict interactions with additional molecules, such as DNA. The BoltzGen team has shown direct binder design is possible for several important use cases such as peptides and small molecules.
Several high performing DNA models have been released in recent years including the Nucleotide Transformer from Instadeep and more recently the Evo models from the Arc Institute. The latter of these demonstrated that their novel striped hyena state-space architecture could be utilised for biological sequences to achieve high performance in a broad set of tasks and maintain long context windows.
Despite these important innovations, no model generalises perfectly outside its training data. The work Lir is doing is right at the edge of existing datasets, meaning none of these tools fit our problem set.
Existing foundation models generalise poorly to novel viral vector design tasks. This is mostly an issue with datasets and model design goals. Viral sequences and structures make up only a small fraction of publicly available datasets; only a small proportion of these are relevant to gene therapy vectors. In our work, we have found this results in poor zero and few shot performance on sequence tasks, along with baseline structure predictions below the standard needed to make useful predictions. In particular, this is a problem if you want to introduce mutations not represented in previous studies and datasets.
Our first target is adeno associated virus (AAV), which is widely used as a gene therapy delivery vector. The majority of the publicly described work on mutating AAV has been limited to small peptide insertional libraries and a couple of well explored mutable regions. Lir is going beyond these.
Generating strong datasets for problems where there are weak priors is challenging. Lab based evolution approaches generally have involved rounds of sampling mostly random variants before moving on to a more targeted screen. There are some advantages to doing things this way. For instance, it allows for relatively low bias in your initial data; however, it has clear downsides of being expensive, slow and potentially yielding low information data.
Many mutations will result in viruses that can’t be produced. This is an initial hurdle before acquiring useful data such as tissue tropism and immune responses. Screening for manufacturable vectors in silico is challenging in the absence of relevant data. Most of Lir’s work involves types of mutations where there is little prior data. This means task specific classifiers and generative models are unlikely to yield good results. Our initial experiments found none of the fully open source foundation models performed well on relevant benchmarks to be suitable for our zero-shot conditions.

LirEvolve is a viral foundation model which is already state-of-the-art in predicting the viability of novel viral vector designs. It is currently being improved further with our own in-house data, with the continued goal of a broadly applicable toolkit for viral vector evolution. This model is allowing Lir to accelerate the work in our lab already. It enables us to run more efficient experiments and focus on generating high quality datasets in every experimental run.
We're taking a holistic, ground up approach to vector engineering. LirEvolve sits alongside our modelling work on protein sequencing, protein folding and beyond, which we'll be talking about here in future updates soon!
LirEvolve is one aspect of our nAAVigator® pipeline which is designed to speed up viral vector design. Combined with our lab-in-the-loop, we’re building a platform to generate vectors specific to clinical partners’ targets at speed. If this sounds relevant to your work please reach out!
References
- ‘Genome Modeling and Design across All Domains of Life with Evo 2’. https://arcinstitute.org/manuscripts/Evo2
- Stark, Hannes, Felix Faltings, MinGyu Choi, et al. ‘BoltzGen: Toward Universal Binder Design’. Preprint, bioRxiv, 24 November 2025. https://doi.org/10.1101/2025.11.20.689494
- Dalla-Torre, Hugo, Liam Gonzalez, Javier Mendoza-Revilla, et al. ‘Nucleotide Transformer: Building and Evaluating Robust Foundation Models for Human Genomics’. Nature Methods 22, no. 2 (2025): 287–97. https://doi.org/10.1038/s41592-024-02523-z- Hayes, Thomas, Roshan Rao, Halil Akin, et al. ‘Simulating 500 Million Years of Evolution with a Language Model’. Preprint, 2 July 2024. https://doi.org/10.1101/2024.07.01.600583
- Notin, Pascal, Aaron Kollasch, Daniel Ritter, et al. ‘ProteinGym: Large-Scale Benchmarks for Protein Fitness Prediction and Design’. Advances in Neural Information Processing Systems 36 (December 2023): 64331–79.
© 2025 Copyright: Lir Therapeutics